
About MPS VII | Rare, Lifelong, & Progressive
MPS VII is a rare, lifelong, and progressive disease
MPS VII is caused by genetic mutations in the GUSB gene
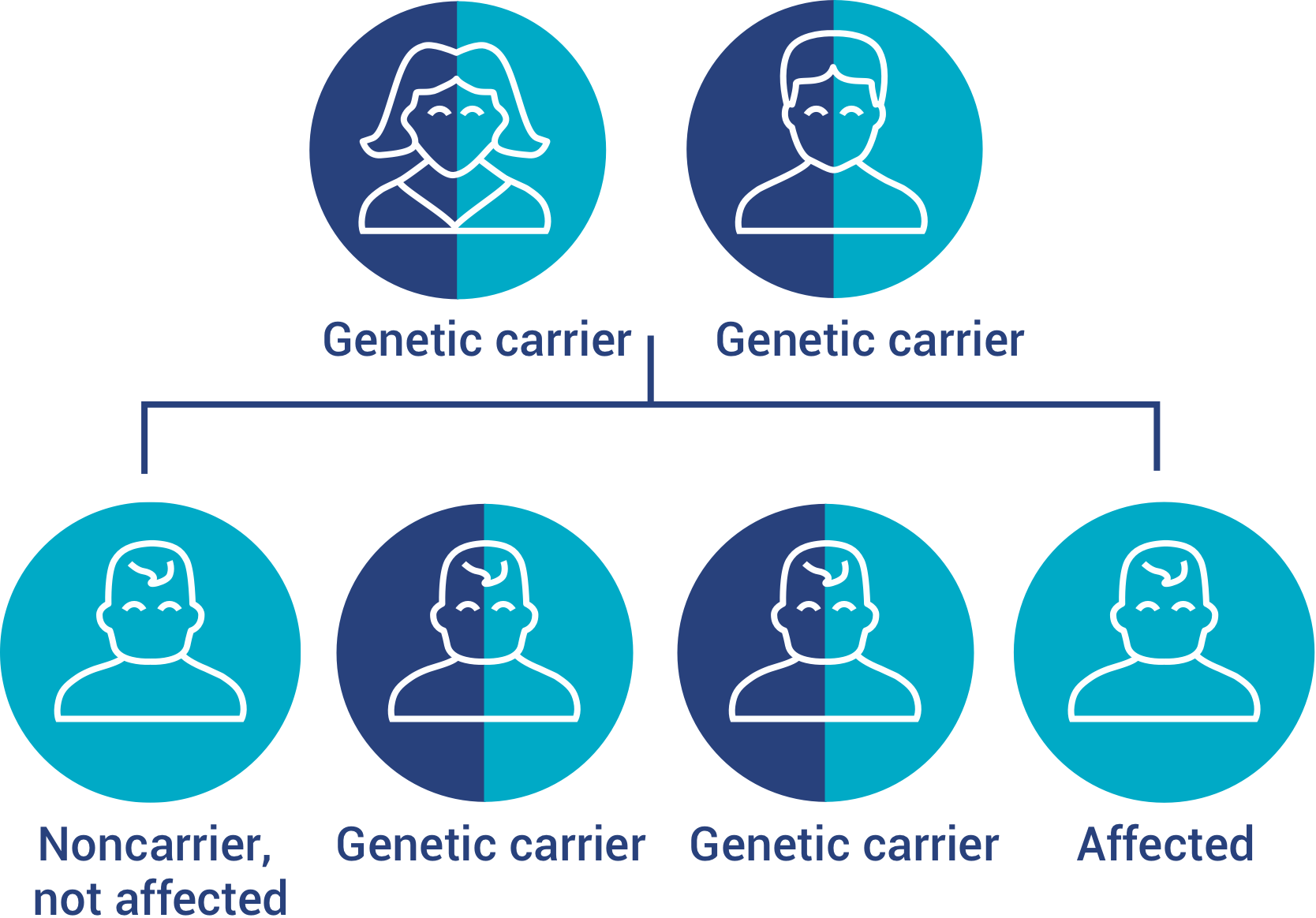
MPS VII is inherited in an autosomal recessive manner
The mutated GUSB gene results in a deficiency in ß-glucuronidase (GUS)—an enzyme that plays a key role in the breakdown of glycosaminoglycans (GAGs).
If both parents are carriers of the mutated GUSB gene, there is a 1 in 4 chance that their child will be born with MPS VII.
- There are approximately 200 people worldwide living with MPS VII
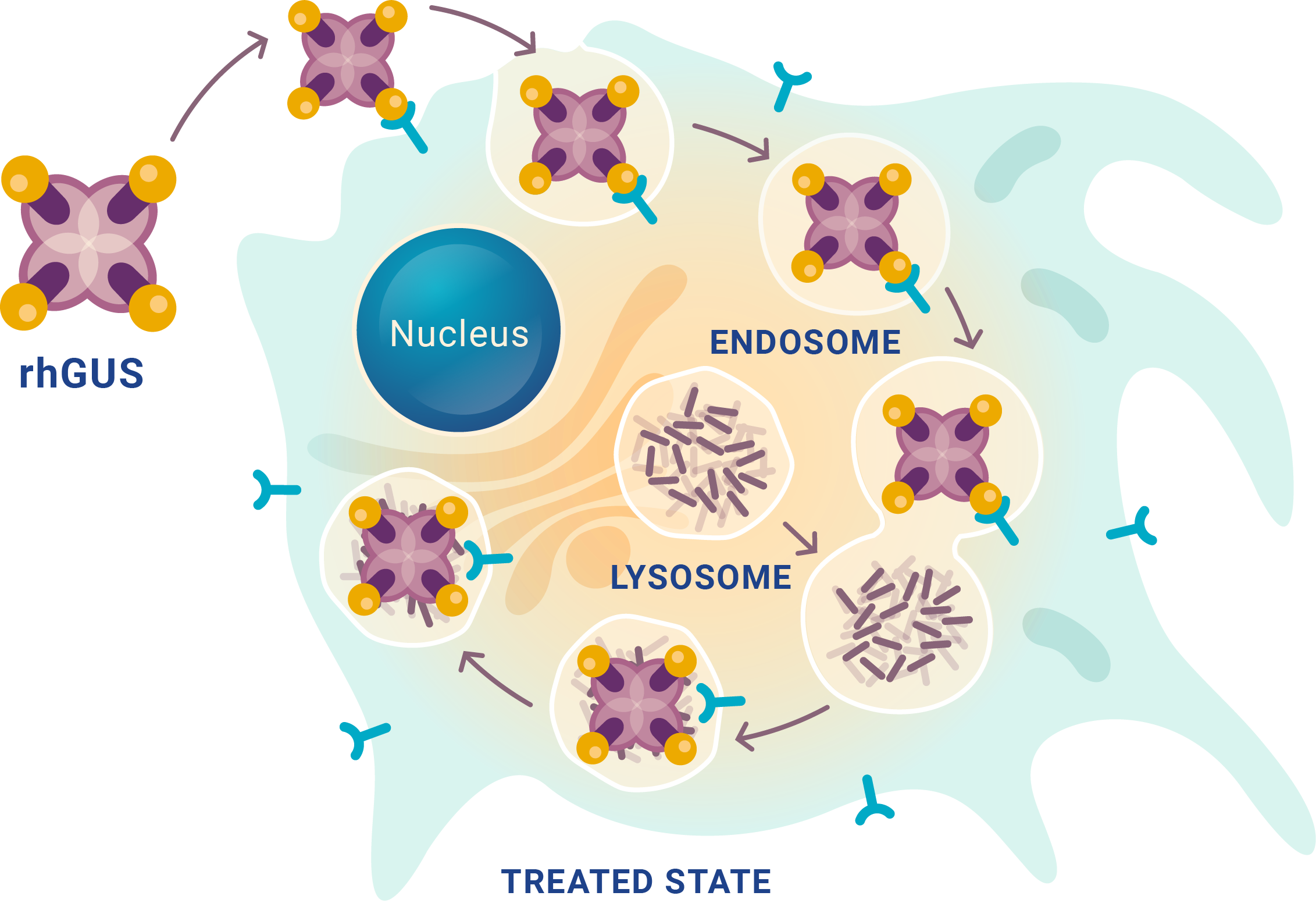
MPS VII is caused by an enzyme deficiency leaving partially degraded glycosaminoglycans (GAGs)* to accumulate in lysosomes
GAGs are complex polysaccharides that must be degraded in a stepwise process by 11 lysosomal enzymes†
- An enzyme deficiency leaves partially degraded GAGs accumulating in lysosomes
- Each enzyme deficiency causes a specific MPS type
- Clinical manifestations are related, but unique to the MPS type
- Excess GAG (dermatan sulfate, chondroitin sulfate, and heparan sulfate) in urine is a marker for MPS
- MPS VII is the only MPS caused by a deficiency of the lysosomal enzyme, ß-glucuronidase (GUS)
*GAGs are a type of mucopolysaccharide—sugars made in the body that help build tissues such as bones, cartilage, skin, and tendons.
†Ten enzymes hydrolyze GAG linkages sequentially; 1 degrades GAG via an alternative route.
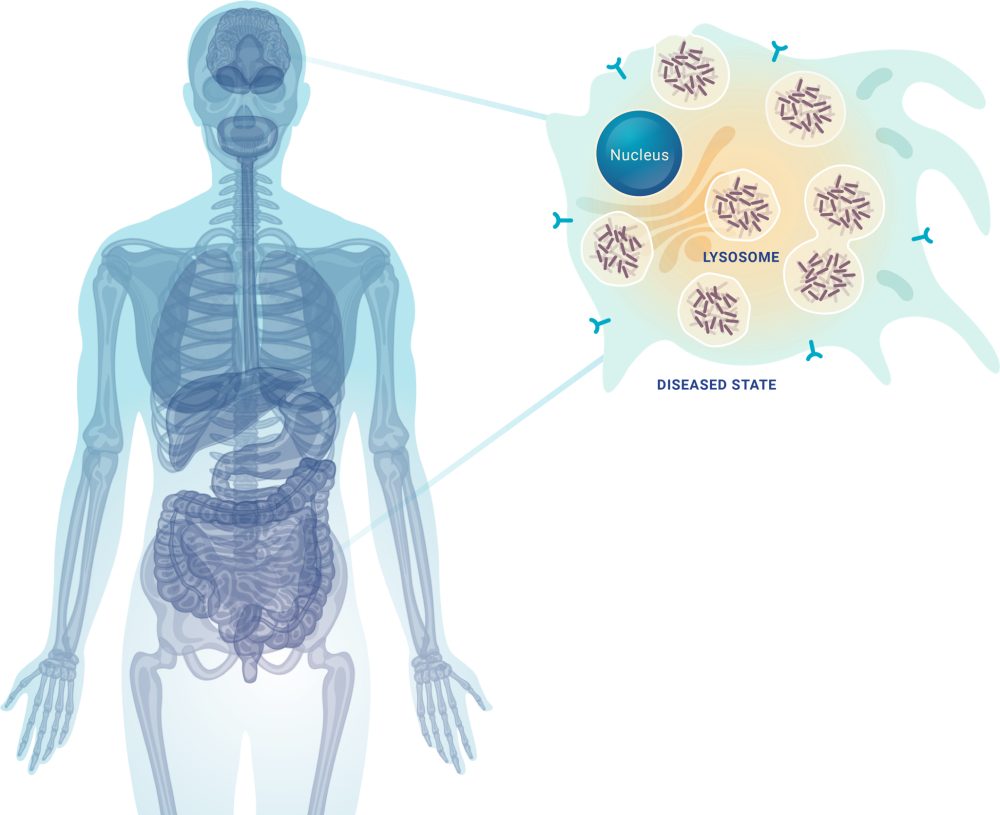
GAG accumulates in lysosomes of cells in tissue, organs, and systems throughout the body
MPS VII manifestations can include:
- Multi-organ dysfunction
- Cognitive impairment
- Reduced life expectancy
MPS VII is progressive
Signs and symptoms of MPS VII generally become more severe over time
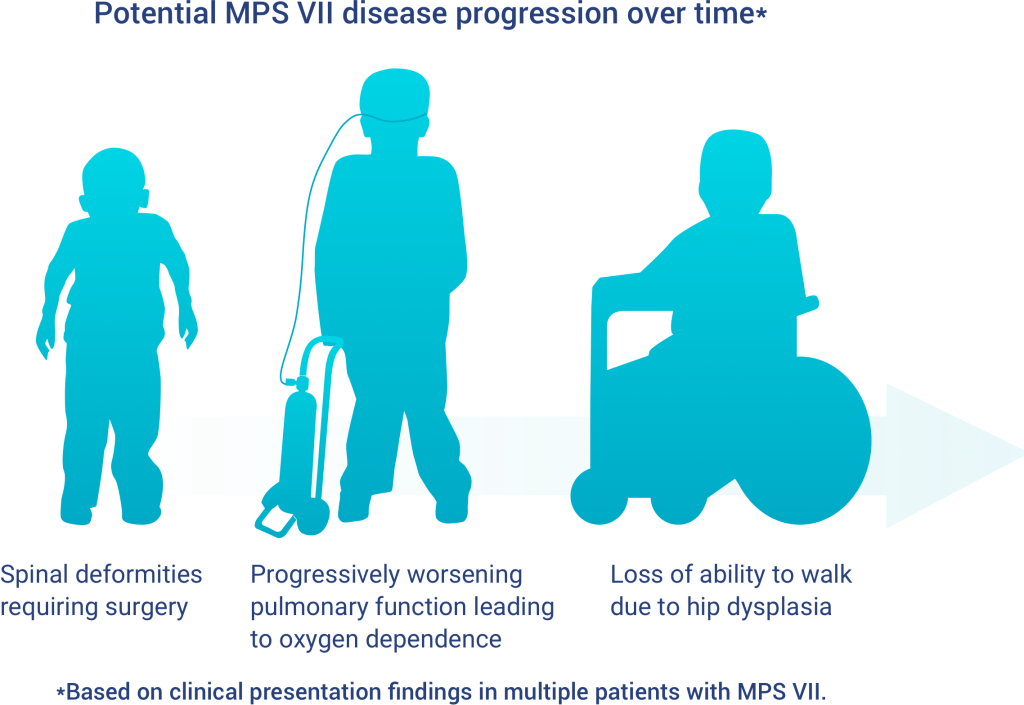
Dysplasia and worsening pulmonary function are progressive symptoms commonly observed in patients with MPS VII. In some cases, people with MPS VII may develop new signs and symptoms which may also progress over time.